

ICU Physiology in 1000 Words: Blood Pressure

Jon-Emile S. Kenny MD [@heart_lung]
Why is blood pressure measured by units of length [i.e. millimeters of mercury – mmHg]? What decides systolic and diastolic pressure? What are the determinants of tissue perfusion and what are the clinical implications of this physiology? Can we deceive ourselves into thinking a particular mean arterial pressure [MAP] is beneficial when, in fact, it is depriving tissue of life? In this brief dispatch, I hope to provide foundation for the above – reasons to reexamine and remarkable references [1-5] for further reading around the omnipresent clinical metric with which we are so familiar.
First Principles
Pressure is a force deployed over an area. For example, the force of one’s body [one’s mass times its gravitational acceleration towards the centre of the Earth – also known as weight] can apply more or less pressure to a snow bank depending upon the area of one’s footwear – the premise behind snow shoes. Historically, when the force of blood applied across the area of the central arteries [i.e. blood pressure] was transduced to a manometer, a mass of mercury [i.e. mercury’s density multiplied its volume] moved up the height of a column – measured in millimeters. One may recall that pressure sensed in this manner is equal to the product of the density of the fluid, gravitational acceleration and height achieved [i.e. pressure = pgh]. Because gravitational acceleration is part of the equation, the elevation at which the pressure is transduced, matters; however – like many of my physiological expositions – the clinical relevance is small. Blood pressure measured by mercury manometry at the peak of Mount Everest is only reduced by 0.2% [3].
Systole & Diastole
Peak systolic pressure is determined by the peak blood volume present in the central arteries during systole and the compliance of the vessels. Importantly, vascular compliance decreases with age and total volume. In other words, at a high baseline blood volume, ejection of additional blood results in greater change in pressure than if the same volume entered the central arteries at a lower baseline volume [3] [see figure 1A]. This physiology is accentuated with each decade of life as the vessels become stiffer [6].
The diastolic pressure is determined by the rate of pressure decay in the central arteries and the time allowed for diastole to transpire. For example, a rapid decay time [i.e. short time constant] combined with a long diastole will favour a low diastolic pressure and vice versa. But what is a time constant of the central arteries? If we assume that the decay from peak systole is mono-exponential, it takes roughly 5 time constants for a distensible structure to return to its baseline state of deformation. A time constant is the product of compliance and downstream resistance. Therefore, a poorly compliant aorta [e.g. old, unhealthy, high volume], coupled with a low downstream resistance [e.g. vasodilator, sepsis, high metabolic demands] will radically reduce the decay-time from systole to diastole. If this is coupled with a long duration of diastole [e.g. beta-blocker], then the final diastolic pressure can be quite low [see figure 1B].
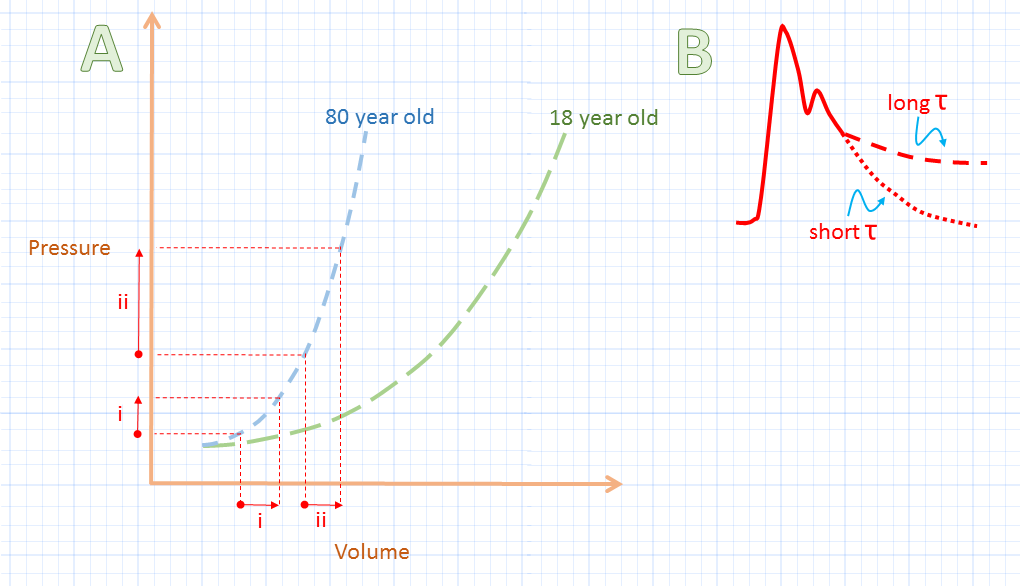
Figure 1A: Volume [x-axis] and flow [assuming stable heart rate] may be the same [compare hypothetical stroke volume i to ii], yet change in pulse pressure from diastole to systole along the y-axis is different because of vascular compliance. This has implications for calculated vascular resistance [see text]. 1B illustrates the difference between long and short time-constant physiology [tau (t) is time constant].
An important clinical implication of the above is that of calculated ‘vascular resistance’; this has been covered previously in the context of pulmonary vascular resistance [7-9]. True downstream resistance may be stable, or even decrease, but if the compliance of the central arteries worsens, then the systolic blood pressure, the mean arterial pressure [MAP] and, consequently, the calculated vascular resistance will rise for a stable flow. This was demonstrated in a canine study where the aorta was reinforced with plastic [decreased compliance] [10]. While there was no change in downstream arteriolar constriction [i.e. ‘true’ resistance], the calculated vascular resistance rose by 20% because of the stiffened aorta. Clinically, an elderly, overloaded patient with a high heart rate could ‘congest’ the aorta and result in an acutely stiffened central vascular space.
Determinant not an Indicator
Blood pressure is a determinant but not an indicator of tissue perfusion [2, 3]. What does this mean? While targeting a specific MAP is recommended in sepsis and septic shock – this may represent a form of ‘tangible bias’ [1, 4, 5]; in other words, we perceive what we can know and measure as more important than it is. Thus, we mistake blood pressure – which we can measure – as an important indication of tissue perfusion, which it is not, per se. If we consider an extreme example – Resuscitative Endovascular Balloon Occlusion of the Aorta [REBOA] – whereby the aorta is occluded by an endovascular balloon in the setting of traumatic blood loss, we can raise the measurable MAP, but vastly diminish tissue perfusion [i.e. everything distal to the balloon].
While the example of REBOA is obviously physiological hyperbole, it serves to remind us that regional organ perfusion is determined not simply by its pressure head [MAP] but also by local resistance – in the setting of REBOA, the local resistance is infinitely high. Increasing MAP at the expense of local perfusion may also be achieved by excessive alpha-receptor agonism which we have all, no doubt, observed in the ICU as skin mottling and/or digital ischemia. How do we reconcile this dissonant clinical scenario where we may be placated by an acceptable MAP yet dispirited by signs of tissue starvation?
As an analogy, consider the quintessential New York City water towers that dot rooftops of the Manhattan skyline. These towers must be filled [e.g. cardiac output] with water and maintain a constant pressure-head [e.g. MAP] for the units in the building [e.g. the tissues]. Some of the units may require large flow – say for manufacturing – and have a low resistance. While other units may require intermittent flow – say for residential – and have relatively high resistance. Accordingly, it is the local demand that determines local resistance and local perfusion. It is expected that cardiac output – the rate of fill of the tower – be independent from the flow in specific units of the building. Increasing flow into the water tank won’t increase the perfusion of a vacant apartment unit! Yet if the water tower runs dry, it is imperative that it be re-filled by increasing its inflow rather than by demanding that all of the building units pinch off their faucets.
In other words, driving up MAP by high-dose vasopressors can be deceptively reassuring. If there is evidence of poor tissue perfusion and if the vital organs are tolerant of further fluid, then augmenting cardiac output with additional volume is physiologically appropriate.
Please check out other articles in this series,
JE
Dr. Kenny is the cofounder and Chief Medical Officer of Flosonics Medical; he is also the creator and author of a free hemodynamic curriculum at heart-lung.org
References
Magder, S., Phenylephrine and tangible bias. Anesthesia & Analgesia, 2011. 113(2): p. 211-213.
Magder, S.A., The highs and lows of blood pressure: toward meaningful clinical targets in patients with shock. Crit Care Med, 2014. 42(5): p. 1241-1251.
Magder, S., The meaning of blood pressure. Critical Care, 2018. 22(1): p. 257.
Thiele, R.H., E.C. Nemergut, and C. Lynch, The physiologic implications of isolated alpha1 adrenergic stimulation. Anesthesia & Analgesia, 2011. 113(2): p. 284-296.
Thiele, R.H., E.C. Nemergut, and C. Lynch, The clinical implications of isolated alpha1 adrenergic stimulation. Anesthesia & Analgesia, 2011. 113(2): p. 297-304.
Nakashima, T. and J. Tanikawa, A study of human aortic distensibility with relation to atherosclerosis and aging. Angiology, 1971. 22(8): p. 477-490.
Naeije, R., et al., The transpulmonary pressure gradient for the diagnosis of pulmonary vascular disease. Eur Respir J, 2013. 41(1): p. 217-23.
Versprille, A., Pulmonary vascular resistance. A meaningless variable. Intensive Care Med, 1984. 10(2): p. 51-3.
Naeije, R., Pulmonary vascular resistance. A meaningless variable? Intensive Care Med, 2003. 29(4): p. 526-9.
Kelly, R.P., R. Tunin, and D. Kass, Effect of reduced aortic compliance on cardiac efficiency and contractile function of in situ canine left ventricle. Circ Res, 1992. 71(3): p. 490-502.