

ICU Physiology in 1000 Words: The Diastolic Shock Index & Subendocardial Perfusion

Jon-Emile S. Kenny MD [@heart_lung]
When we model the circulation at the bedside, we resort to generalizations. While these mechanistic heuristics don’t completely capture the complexity of hemodynamics, they do help us think quickly when confronted by clinical urgency. For example, distilling ‘venous return’ into an upstream pressure and downstream pressure separated by a single resistance, is certainly simplified [1-5]. As well, we often tie together the branching arterial tree into sequential ‘generations’ and assume each division to be completely characterized by a single compliance and resistance. Such an approach ignores the uniqueness of each organ and tissue bed; for example, we know that the skin, brain, kidneys and heart all behave differently at the level of pre-capillary arteriole [6-8]. As previously argued, ‘All Perfusion is Local’ and one way to model each tissue’s distinct perfusion characteristic is through ‘conductance.’
Conductance
Conductance is the inverse of resistance. In a hemodynamic model, a conductance curve is generated with pressure on the x-axis and flow on the y-axis [Figure 1]; the slope of the line, is conductance. While simply the mathematical inverse of resistance, I prefer to use ‘conductance’ because it sheds the cognitive baggage that clings to ‘resistance.’ That is, ‘resistance’ conjures an image of constricting and dilating vessels. No doubt, pre-capillary arteriolar constriction decreases flow per unit pressure and the conductance curve shifts down [8]. But there is more to conductance than cross-sectional vessel area! For example, stiffening a vessel decreases flow per unit pressure. This is why, in an experimental model, reinforcing the aorta with plastic decreases conductance [increases resistance] without any change in vessel diameter [9]. It’s also why the pulmonary vascular circulation can recruit and dilate in response to high pulmonary artery occlusion pressure – yet have diminished conductance [i.e. because the vessels stiffen at their elastic limit] [10].
Conductance & Time
Yet another assumption collides with the aforementioned and it stems from how we calculate flow. Tissue flow [volume per time] is calculated as the product of stroke volume [volume per cycle] and heart rate [cycles per minute]; this is true if, and only if, the tissue bed receives volume throughout the entire cycle [i.e. during systole and diastole]. Accordingly, if tissue preferentially accepts volume during a specific portion of the cardiac cycle, then the direct relationship between flow and cycle rate falls apart. Why? Because as heart rate changes so too does the proportion of systole and diastole [11, 12].
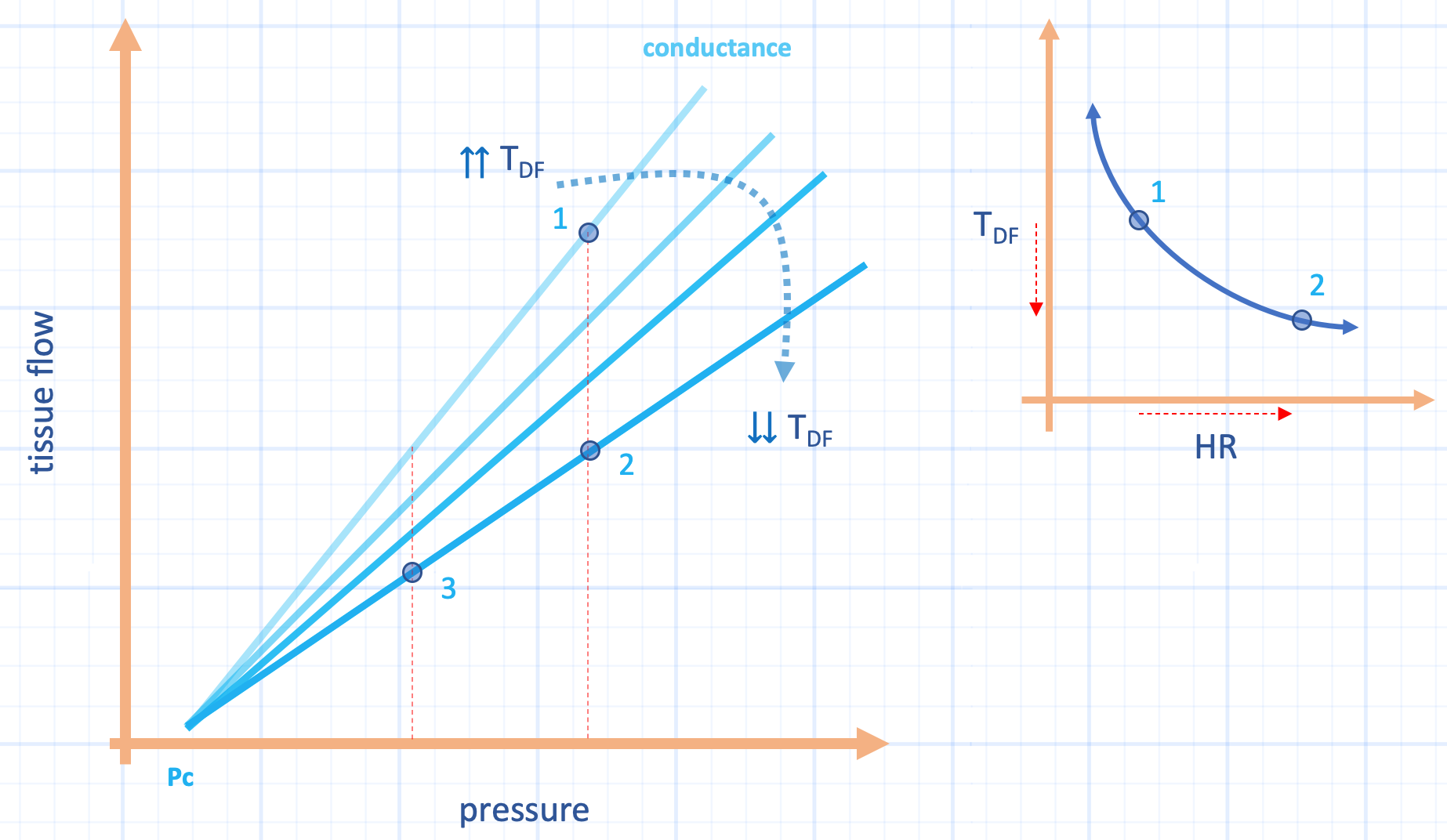
Figure 1: Hypothetical effect of heart rate on diastolic time fraction [Tdf] and conductance. The effect of increasing heart rate [HR, x-axis, inset] from point 1 to 2 on the inset shows large drop in Tdf [y-axis, inset]. This effect decreases subendocardial conductance [slope of light line blue lines]. Thus, tissue flow [y-axis] falls to point 2 secondary to high heart rate. Healthy hearts can blunt the effect of decreased Tdf by vasodilation [which increases conductance] and also, increased heart rate tends to raise diastolic pressure, elevating the x-axis. During septic shock, however, as diastolic pressure falls, especially with high heart rate [i.e. high DSI], perfusion falls to point 3. Pc is closing pressure and assumed constant for clarity.
Consider the subendocardium and the midmyocardium. During systole, blood volume is emptied from these portions of the heart muscle [13-15]; they are re-perfused during cardiac relaxation. In other words, these subcomponents of the myocardium preferentially receive blood flow during diastole. Thus, their perfusion is described by the diastolic pressure gradient [i.e. coronary diastolic pressure less left ventricular diastolic pressure] and the fraction of time spent in diastole [13-15]. Indeed, when coronary perfusion is impaired, its vascular conductance is increased not just by vessel dilation, but also by preferentially prolonging the fraction of time spent in diastole, even when heart rate is held constant [16]! By contrast, the conductance of the subendocardium decreases the most by shortened diastolic fraction compared to the midmyocardium, while the subepicardium is independent of diastolic time fraction [14]. Thus, studies of human septic shock showing increased coronary conductance, blood flow and diminished oxygen extraction [17, 18] could, possibly, be explained by preferential flow through the subepicardium and into the coronary sinus [19]. Increased subepidcardial-to-subendocardial blood flow secondary to heart rate elevation is well-described [15, 20, 21]. This is important because starved subendocardium contributes to cardiac dysfunction [22], which is observed in septic shock.
Implications for Diastolic Shock Index
In a recent retrospective analysis, the diastolic shock index [DSI, the heart rate divided by the diastolic blood pressure] predicted mortality in septic shock; neither heart rate, nor diastolic pressure alone did so [23]. Rightly, the authors argue that rising DSI is an index of septic shock severity. Why? As previously described, the diastolic blood pressure is directly proportional to the arterial time constant and inversely to the duration of the cardiac cycle. The time constant comprises vascular ‘tone’ [e.g. cross-sectional area] and stiffness; thus, vasodilation shortens the time constant and diminishes end-diastolic pressure. If the heart rate is also low [i.e. long cycle time], then the diastolic pressure will fall. By this reasoning, a fast heart rate [i.e. short cycle time] raises the diastolic pressure, by allowing less time for pressure decay. Accordingly, a high DSI [rising heart rate with low diastolic blood pressure] indicates a severely shortened time constant [e.g. vasodilation] as the fast heart rate should pull up the diastolic pressure in the denominator.
While all of the above is true, and noteworthy, the DSI might also be considered a specific indicator of subendocardial perfusion. Heart rate and the fraction of diastole have a negative exponential relationship [Figure 1, inset] [13]. Thus, rising heart rate in the numerator is a surrogate for increasingly short subendocardial perfusion time. As above, this diminishes subendocardial conductance. When coupled with falling diastolic pressure in the denominator, the DSI then describes the midmyocardium and subendocardium at high risk for ischemia.
While the DSI was studied retrospectively and therapeutic implications are premature, it does cast new perspectives on previous investigations. In a recent, un-resuscitated sepsis population, intravenous fluids decreased the DSI primarily by lowering heart rate; interestingly, left ventricular end-systolic elastance increased [suggesting improved inotropic state] [24]. If we assume that end-systolic elastance is a load-independent marker of contractility, then perhaps the fluids improved subendocardial perfusion by raising diastolic time fraction? The DSI also brings to mind less-recent investigations indicating salutary effects for beta-blockade in septic shock.
As always, pre-test probability and patient population is critical, especially when considering therapy. One can imagine an elevated DSI in severe, acute congestive heart failure with aortic regurgitation; clearly, intravenous fluid to lower DSI would be a wrongheaded approach.
Best,
JE
Dr. Kenny is the cofounder and Chief Medical Officer of Flosonics Medical; he also the creator and author of a free hemodynamic curriculum at heart-lung.org. Download his free textbook here.
References
Gelman S: Classic Papers Revisited: My Love Affair with the Venous System. Anesthesiology: The Journal of the American Society of Anesthesiologists 2018, 129(2):329-332.
Gelman S, Bigatello L: The physiologic basis for goal-directed hemodynamic and fluid therapy: the pivotal role of the venous circulation. Canadian Journal of Anesthesia/Journal canadien d'anesthésie 2018, 65(3):294-308.
Gelman S: Venous function and central venous pressure: a physiologic story. Anesthesiology 2008, 108(4):735-748.
Magder S, Scharf SM: Venous return; Magder, Sheldon, and Steven M. Scharf. "Venous return." Lung Biology in Health and Disease 157 (2001): 93-112.
Magder S: Volume and its relationship to cardiac output and venous return. Critical care 2016, 20(1):271.
Magder SA: The highs and lows of blood pressure: toward meaningful clinical targets in patients with shock. Critical care medicine 2014, 42(5):1241-1251.
Magder S: The meaning of blood pressure. Critical Care 2018, 22(1):257.
Magder S: Pressure-flow relations of diaphragm and vital organs with nitroprusside-induced vasodilatation. Journal of Applied Physiology 1986, 61(2):409-416.
Kelly RP, Ting C-T, Yang T-M et al: Effective arterial elastance as index of arterial vascular load in humans. Circulation 1992, 86(2):513-521.
Tedford RJ: Determinants of right ventricular afterload (2013 Grover Conference series). Pulmonary circulation 2014, 4(2):211-219.
Chung CS, Karamanoglu M, Kovacs SJ: Duration of diastole and its phases as a function of heart rate during supine bicycle exercise. American Journal of Physiology-Heart and Circulatory Physiology 2004, 287(5):H2003-H2008.
Bombardini T, Gemignani V, Bianchini E et al: Diastolic time–frequency relation in the stress echo lab: filling timing and flow at different heart rates. Cardiovascular ultrasound 2008, 6(1):15.
Boudoulas H, Rittgers SE, Lewis R et al: Changes in diastolic time with various pharmacologic agents: implication for myocardial perfusion. Circulation 1979, 60(1):164-169.
Fokkema DS, VanTeeffelen JW, Dekker S et al: Diastolic time fraction as a determinant of subendocardial perfusion. American Journal of Physiology-Heart and Circulatory Physiology 2005.
Ferro G, Duilio C, Spinelli L et al: Relation between diastolic perfusion time and coronary artery stenosis during stress-induced myocardial ischemia. Circulation 1995, 92(3):342-347.
Merkus D, Kajiya F, Vink H et al: Prolonged diastolic time fraction protects myocardial perfusion when coronary blood flow is reduced. Circulation 1999, 100(1):75-81.
Cunnion RE, Schaer GL, Parker MM et al: The coronary circulation in human septic shock. Circulation 1986, 73(4):637-644.
Dhainaut J-FO, Huyghebaert M-F, Monsallier J et al: Coronary hemodynamics and myocardial metabolism of lactate, free fatty acids, glucose, and ketones in patients with septic shock. Circulation 1987, 75(3):533-541.
Adiseshiah M, Baird RJ: Correlation of the changes in diastolic myocardial tissue pressure and regional coronary blood flow in hemorrhagic and endotoxic shock. Journal of Surgical Research 1978, 24(1):20-25.
Buffington CW, Sivarajan M, Bashein G: The quotient of mean arterial pressure and heart rate predicts hypoperfusion of collateral-dependent myocardium. Journal of cardiothoracic anesthesia 1989, 3(1):65-69.
Buckberg GD, Fixler DE, Archie JP et al: Experimental subendocardial ischemia in dogs with normal coronary arteries. Circulation research 1972, 30(1):67-81.
Ross Jr J: Mechanisms of regional ischemia and antianginal drug action during exercise. Progress in cardiovascular diseases 1989, 31(6):455-466.
Ospina-Tascón GA, Teboul J-L, Hernandez G et al: Diastolic shock index and clinical outcomes in patients with septic shock. Annals of intensive care 2020, 10:1-11.
Guarracino F, Bertini P, Pinsky MR: Cardiovascular determinants of resuscitation from sepsis and septic shock. Critical Care 2019, 23(1):118.