

ICU Physiology in 1,000 Words: ARDS - Part 3

Jon-Emile S. Kenny [@heart_lung]
While parts 1 and 2 of this trilogy considered the mechanical power applied to the lung skeleton and the effects of lung inhomogeneity [i.e. ‘stress raisers’], respectively; this final installment will draw the reader towards the pulmonary vasculature as a key mediator of ventilator induced lung injury [VILI]. That the pulmonary vasculature participates in the pathogenesis and propagation of the acute respiratory distress syndrome [ARDS] has been known for decades [1]. Highlighting the importance of the pulmonary vascular tree in ARDS, both a prediction score for acute cor pulmonale [ACP] in ARDS and an expert opinion on the hemodynamic management of ARDS have been – very recently – circulated [2, 3].
Blood Vessels and VILI
There are a plethora of ‘indirect’ insults to the pulmonary vasculature which raise resistance to blood flow [4, 5] during ARDS. These include microthrombi, hypoxemic vasoconstriction, hyperpcapnia and interstitial edema [4]. Additionally, the ‘direct’ effect of augmented lung stress and airway mechanics upon the vasculature are also paramount [5]. In a patient, fully adapted with the ventilator [e.g. paralyzed], a mechanical breath raises lung stress as detailed in part 1. Stress on the lung skeleton has opposing effects upon the two vascular populations within the lung [6]. The alveolar vessels [e.g. the pulmonary capillaries] are exposed to the alveolar pressure and tend to collapse during lung inflation [i.e. because the alveolar pressure rises relative to the pleural pressure]. By contrast, within the bronchovascular bundles – which originate at the hila and invaginate into the lung parenchyma – lie the extra-alveolar vessels. When lung volume rises [even in response to a fully passive, ventilator-delivered breath] these conduits dilate. To the extent that these conduits increase in size, the interstitial pressure surrounding the extra-alveolar vessels falls; consequently, the extra-alveolar vessels tend to dilate and elongate [7].
There is a unique sub-population of blood vessels within the lung known as ‘corner vessels’ which are anatomically alveolar [they lie at the junction of at least 3 alveolar septae] but are physiologically extra-alveolar because they dilate when lung volume rises [6]. Indeed, under West zone I conditions, the corner vessels are responsible for continued, local blood flow [8]. When lung stress is abnormally high – as during ARDS – over-distention of inhomogeneous lung will result in excessive West Zone I, particularly during inflation. This will force excess blood through corner vessels, raise their transmural pressure and favor interstitial edema, particularly when vessels are abnormally leaky as in ARDS [3, 6].
While the Mead model detailed in part 2 discusses the effects of stress raisers on neighboring lung units during inflation, asymmetrical radial forces are exerted in an equal, but opposite direction during exhalation [9]. Accordingly, the interstitial space adjacent to a stress raiser during alveolar emptying is predicted to experience traction forces multiplied by a factor of 4.5 [6, 9]. Thus, if alveoli begin to empty at an end-inspiratory pressure of 30 cm H2O, the interstitial pressure surrounding the corner vessel may fall by 140 cm H2O [or 100 mmHg]! It follows that the transmural pressure of the corner vessel can – in theory – raise to a level known to fracture capillaries [10] [see figure 1].
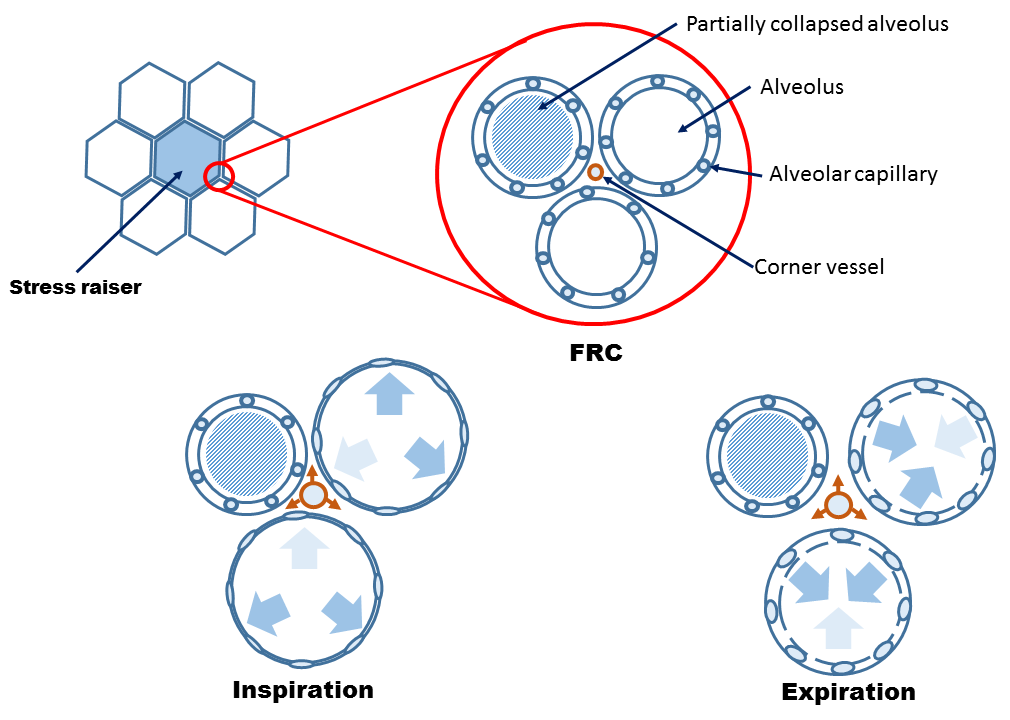
Figure 1: Cartoon demonstrating Mead model [9] of asymmetrical radial traction forces placed upon alveoli and pulmonary vasculature. See Part 2 for definition of stress raiser. Inspiration collapses alveolar capillaries which shunts blood through corner vessels [orange vessel]. Expiration, like inspiration results in asymmetrical traction forces as alveoli shrink, resulting in further traction on corner vessels. FRC is functional residual capacity
In summation, the mechanical power applied to the airways can exacerbate vascular injury via cyclical stress on the microvasculature. Empirically, increased pulmonary arterial blood flow, cycle time and pressure have all been demonstrated to exacerbate hemorrhagic edema and VILI [6, 11-14]. Additionally, in a VILI model, lowering left atrial pressure exacerbates lung injury [15]. From above, it may be deduced that diminished left atrial and pulmonary venous pressure will increase the fraction of West zone I and II thereby exacerbating cyclical fluctuation in microvascular stress. By this reasoning, West zone III acts – in some ways – as ‘vascular PEEP.’
Monitoring ACP
In response to the aforementioned vascular injury – and consequent high right ventricular [RV] afterload – the RV may suffer from dilation [i.e. diastolic dysfunction or volume overload] and paradoxical septal motion [i.e. systolic dysfunction or pressure overload]. When both RV diastolic and systolic dysfunction are present on an echocardiogram, ACP is diagnosed [16]. Prior to lung protective ventilation [LPV], the incidence of ACP in ARDS was upwards of 60%, while in contemporary studies, it is about 20% [5]. Fortunately, clinicians can now better predict the likelihood of ACP based on a recent evaluation [2] [see table 1]
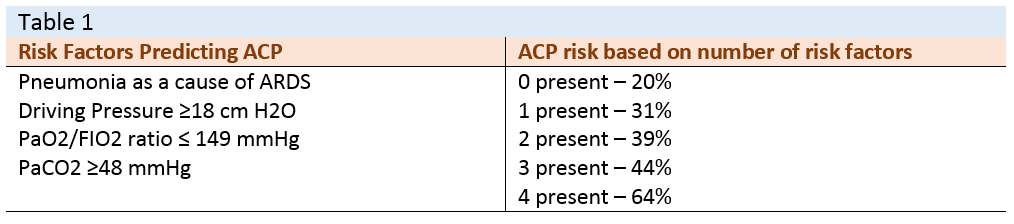
[see reference [2]]
Non-invasive indices of fluid responsiveness in ARDS pose problems as RV output may vary in response to respiratory-induced augmentation in afterload, rather than decrement in preload [17]. The use of trans-esophageal echocardiography may mitigate such error by continuous assessment of RV size, SVC collapsibility [i.e. in the paralyzed patient] and RV outflow mean acceleration time [17]. The latter index is the ratio of the maximal velocity of RV outflow to the acceleration time; its fall is directly associated with RV systolic function and inversely related to RV afterload [18].
Another venerable monitoring utensil is the pulmonary artery catheter [PAC]. Importantly, because of the near-ubiquitous presence of tricuspid regurgitation during mechanical ventilation [19] [especially in ARDS [20]], thermodilution cardiac output should be abandoned. Similarly, pulmonary vascular resistance is likely a meaningless variable [21, 22]. Instead, untoward hemodynamic effects of mechanical ventilation upon the RV may be inferred from diminished pulmonary arterial pulse pressure and/or increased isovolumic contraction pressure [see figure 2] [23].
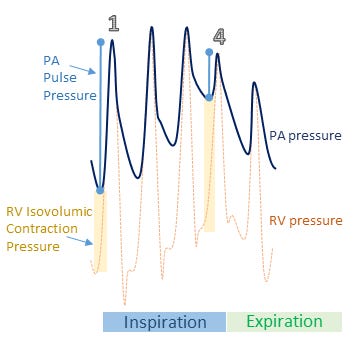
Figure 2: Demonstration of inspiratory embarrassment of RV function by alveolar over-distention [e.g. stress index > 1]. Beat 1 [onset of mechanical inspiration] has relatively low isovolumic contraction pressure [vertical light yellow shaded bar] which indicates the pressure required by the RV to open the pulmonic valve. The PA pulse pressure [light blue dumbbell] is relatively large, indicating a large RV stroke volume. By contrast, beat 4 has a large isovolumic contraction pressure and low pulse pressure indicating a large pressure gradient required by the RV to eject and a low RV stroke volume, respectively. This means of monitoring requires simultaneous PA pressure [dark blue] and RV pressure [dotted yellow] tracings. See reference [23]
Therapy
Ultimately, the above underscores the importance of LPV; adhering to the adage ‘what’s good for the lung is good for the RV’ [20] certainly applies. Selection of optimal PEEP using driving pressure or the stress index as quantitative and qualitative surrogates – respectively – of alveolar distention, minimizing mechanical power applied to the lung, prone positioning, the use of norepinephrine to maintain right coronary artery perfusion in shocked ARDS patients as well as the judicious use of fluids are all initial considerations. Further, deliberation for pulmonary arterial vasodilators and extra-corporeal support should be part of one’s hemodynamic approach to severe ARDS [3, 5]. Nevertheless, what remains unanswered is whether specifically monitoring and treating ACP in ARDS will improve outcome.
Best,
Zapol, W.M. and M.T. Snider, Pulmonary hypertension in severe acute respiratory failure. New England Journal of Medicine, 1977. 296(9): p. 476-480.
Dessap, A.M., et al., Acute cor pulmonale during protective ventilation for acute respiratory distress syndrome: prevalence, predictors, and clinical impact. Intensive Care Med, 2016: p. 1-9.
Vieillard-Baron, A., et al., Experts’ opinion on management of hemodynamics in ARDS patients: focus on the effects of mechanical ventilation. Intensive Care Med, 2016. 42(5): p. 739-749.
Guérin, C. and M.A. Matthay, Acute cor pulmonale and the acute respiratory distress syndrome. Intensive Care Med, 2016: p. 1-3.
Repessé, X., C. Charron, and A. Vieillard-Baron, Acute respiratory distress syndrome: the heart side of the moon. Curr Opin Crit Care, 2016. 22(1): p. 38-44.
Marini, J.J., J.R. Hotchkiss, and A.F. Broccard, Bench-to-bedside review: microvascular and airspace linkage in ventilator-induced lung injury. Critical Care, 2003. 7(6): p. 435.
Goshy, M., S.J. Lai-Fook, and R.E. Hyatt, Perivascular pressure measurements by wick-catheter technique in isolated dog lobes. J Appl Physiol, 1979. 46(5): p. 950-955.
Lamm, W., et al., Flow through zone 1 lungs utilizes alveolar corner vessels. J Appl Physiol, 1991. 70(4): p. 1518-1523.
Mead, J., T. Takishima, and D. Leith, Stress distribution in lungs: a model of pulmonary elasticity. J Appl Physiol, 1970. 28(5): p. 596-608.
Mathieu-Costello, O., et al., Pulmonary capillaries are more resistant to stress failure in dogs than in rabbits. J Appl Physiol, 1995. 79(3): p. 908-917.
Broccard, A.F., et al., Consequences of vascular flow on lung injury induced by mechanical ventilation. Am J Respir Crit Care Med, 1998. 157(6): p. 1935-1942.
Broccard, A.F., et al., Effects of mean airway pressure and tidal excursion on lung injury induced by mechanical ventilation in an isolated perfused rabbit lung model. Crit Care Med, 1999. 27(8): p. 1533-1541.
Hotchkiss Jr, J.R., et al., Effects of decreased respiratory frequency on ventilator-induced lung injury. Am J Respir Crit Care Med, 2000. 161(2): p. 463-468.
Hotchkiss Jr, J.R., et al., Relative roles of vascular and airspace pressures in ventilator-induced lung injury. Crit Care Med, 2001. 29(8): p. 1593.
Broccard, A.F., et al., Impact of low pulmonary vascular pressure on ventilator-induced lung injury*. Crit Care Med, 2002. 30(10): p. 2183-2190.
Jardin, F., O. Dubourg, and J.-P. Bourdarias, Echocardiographic pattern of acute cor pulmonale. CHEST Journal, 1997. 111(1): p. 209-217.
Repessé, X., C. Charron, and A. Vieillard-Baron, Assessment of the effects of inspiratory load on right ventricular function. Curr Opin Crit Care, 2016. 22(3): p. 254-259.
Vieillard-Baron, A., et al., Cyclic changes in right ventricular output impedance during mechanical ventilation. J Appl Physiol, 1999. 87(5): p. 1644-1650.
Jullien, T., et al., Incidence of tricuspid regurgitation and vena caval backward flow in mechanically ventilated patients: a color Doppler and contrast echocardiographic study. CHEST Journal, 1995. 107(2): p. 488-493.
Repessé, X., C. Charron, and A. Vieillard-Baron, Acute cor pulmonale in ARDS: rationale for protecting the right ventricle. CHEST Journal, 2015. 147(1): p. 259-265.
Versprille, A., Pulmonary vascular resistance. A meaningless variable. Intensive Care Med, 1984. 10(2): p. 51-3.
Naeije, R., Pulmonary vascular resistance. A meaningless variable? Intensive Care Med, 2003. 29(4): p. 526-9.
Jardin, F., et al., Relation between transpulmonary pressure and right ventricular isovolumetric pressure change during respiratory support. Catheterization and cardiovascular diagnosis, 1989. 16(4): p. 215-220.